It is possible to interact with the Discover repositories programmatically by using DiscoverPy- a python package developed by us. DiscoverPy comes pre-installed in all Polly notebooks. As of now, it can be used only in a Polly environment.
2.2.1 Elastic search indices in Discover
According to the FAIR’s Findable principle, (Meta)data should be indexed in a searchable resource so that we can find a record with its unique identifier or any of its rich metadata. For that, we’ve used Elasticsearch and put repositories data and metadata in Elasticsearch Indexes.
We’ve also provided APIs over HTTPS with authentication for accessing the data in a secured manner which aligns with the FAIR’s Accessible principle of (Meta)data should be retrievable by their unique identifier using a standardized communications protocol.
2.2.2 Discoverpy Usage
2.2.2.1 Initialisation
- We need a discover object to interact with a data repository in Discover.
from discoverpy import Discover
discover = Discover()
discover- Each data repository on Polly has a unique ID that is used to refer to that repository. The following command list the available data repositories, their ID, and other information associated with them.
discover.get_repositories()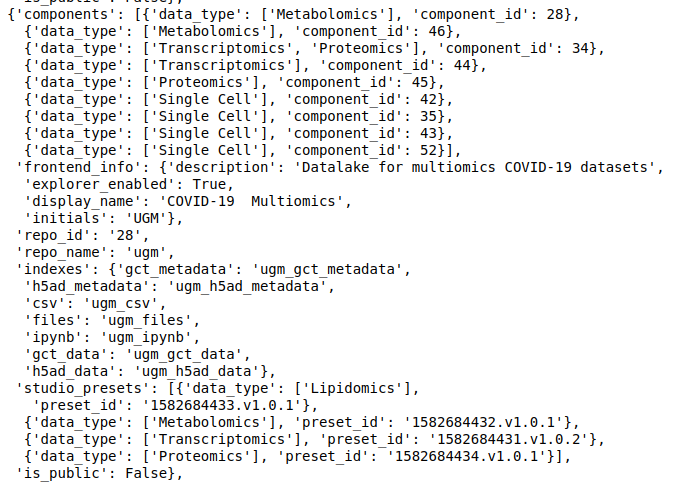
- Next, we need to set the context to a repository, so that all the subsequent queries will be run against that repository. There is another parameter mode, that we can set for
single-cell repositories mode= single_cell, which will search across cells.
bulk data repositories mode= bulk, which will search across samples (default).
-
Set a repository for the discover object You can use the id of a repository to set the discover object to point to that repository.
-
For single cell repositories use
mode='single_cell'. - For bulk data repositories use
mode='bulk'(default)
For instance, the GEO repository has repo_id 9.
discover.set_repo('9')The single cell repository's repo_id is 17.
discover.set_repo("17", mode="single_cell")After you’ve added the indices for a repository, you can view the discover object. It lists the various indices associated with the repository set.
discover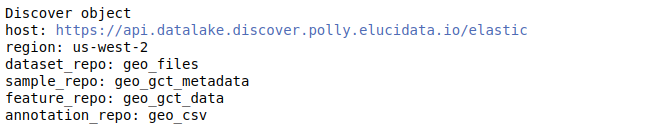
Understanding the Discover object The discover object contains different slots, each containing different levels of curated information. While querying using the Discover object, we need to specify a slot explicitly - which denotes the level at which we want to query. Thus, it is essential to know what information each slot contains-
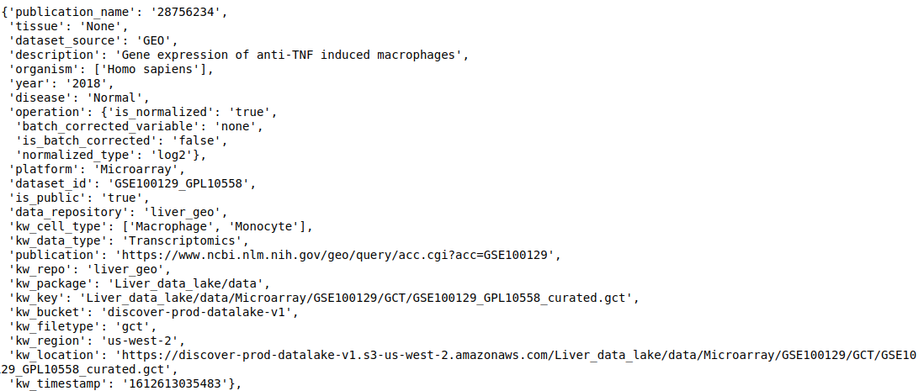
- dataset_repo - This slot contains the study level metadata which consists of identifiers for diseases, cell lines, tissue, drugs, organism, and cell types. The identifier maps to concepts in biomedical ontologies and is consistent across datasets. Refer to Dataset level annotations for more information about the curation at the dataset level.
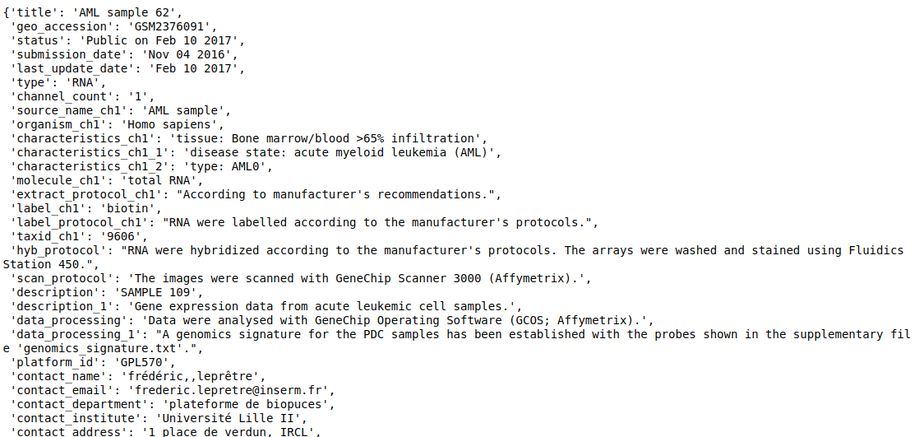
- sample_repo - This slot contains sample level metadata such as genotype, cell lines, tissue, drugs, siRNA, etc., and is also tagged using ontologies. For multi-sample datasets, it also contains the variable which informs about whether the sample is a control sample or a perturbation sample in the context of the study. With this, we should be able to run queries that find metastatic and primary tumors, finds samples treated with a particular drug, or more generally, look for a sample that has a specific phenotype. Refer to Sample level annotation for more information about the curation at the sample level.
- feature_repo - This is the slot containing matrix data of a dataset that contains the rows (feature identifiers) and the actual measurement value for those features for the samples in the dataset. The features could be anything depending on the type of study. For example, for transcriptomics datasets, the feature identifiers will be genes(HGNC symbols) and for proteomics datasets, they will be Proteins(referring back to UNIPROT).
- annotation_repo - With this slot, we can access various feature annotation databases to get information about a particular feature or a set of features.
2.2.2.2 Querying
As our curation process stores metadata values under a controlled vocabulary, the queries in discoverpy follow FAIR's Interoperability guideline. This means that all the users' queries are searched against keywords in a search space that is limited to the vocabulary present in biological ontologies.
2.2.2.2.1 Common methods for multiple indices.
There are methods available on all levels of repository indices. You just need to replace <INDEX_LEVEL_NAME> with one of sample_repo | feature_repo | dataset_repo | annotation_repo
- Get fields present in the index.
discover.<INDEX_LEVEL_NAME>.get_all_fields()For example, to get the fields present for the datasets in this repository,
discover.dataset_repo.get_all_fields()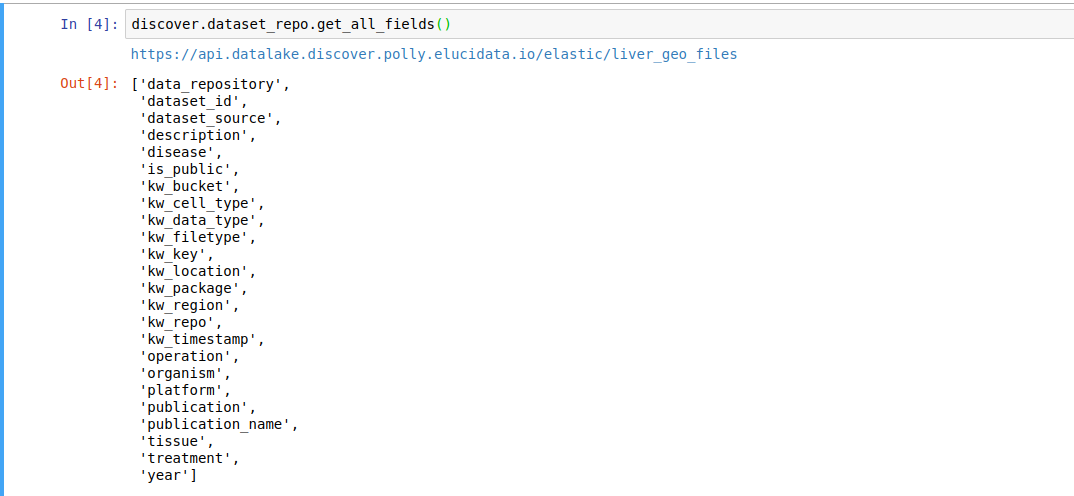
- Fetch the top
nrecords from a repository.discover.<INDEX_LEVEL_NAME>.get_n_records()
For example, to get the fields present for datasets in this repository,
discover.dataset_repo.get_all_fields()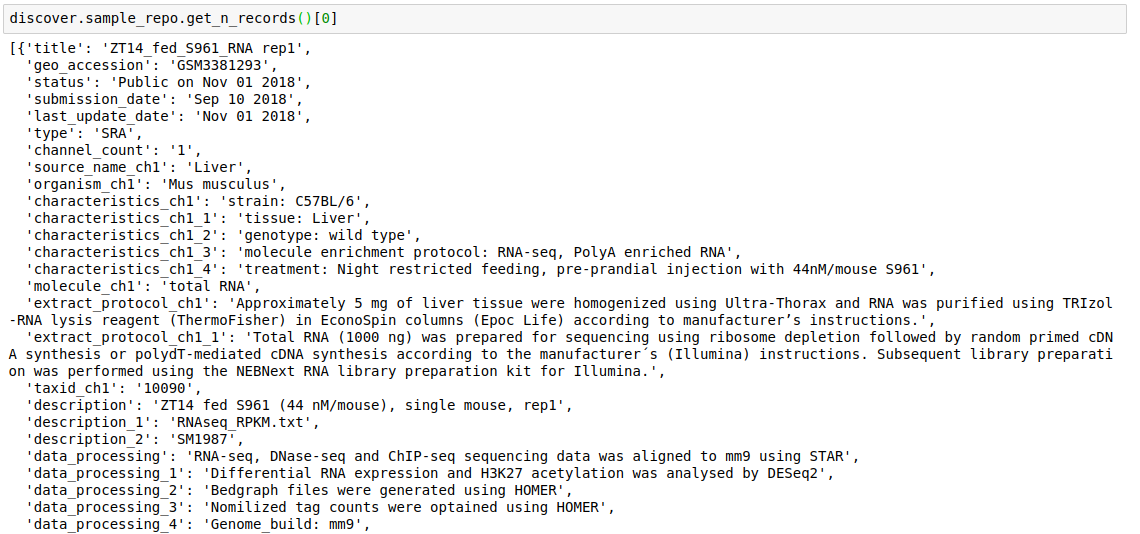
- Output the records as a dataframe -
discover.<INDEX_LEVEL_NAME>.get_n_records_as_df()[0]
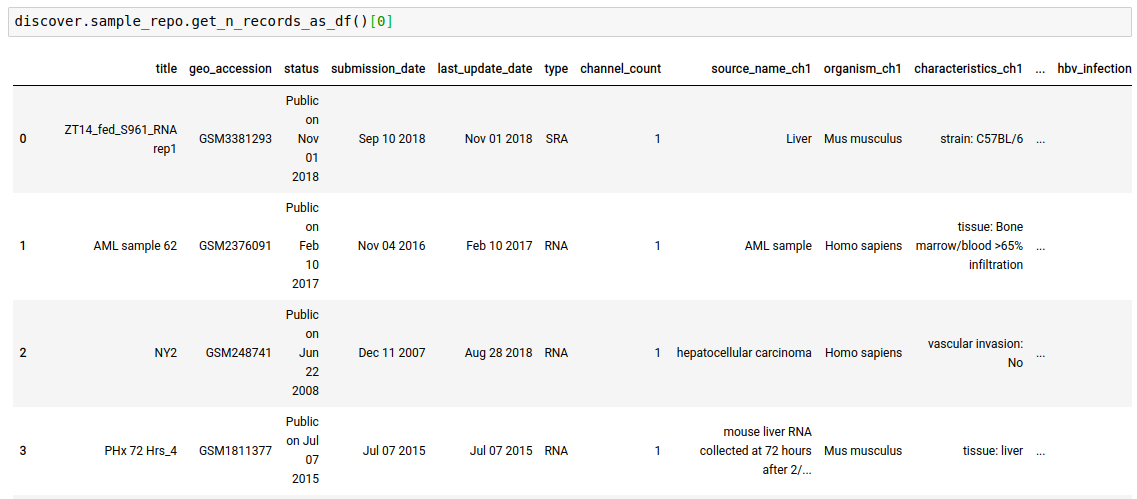
-
Note: You would find there are many methods that have the *_as_df variant as well, which takes the same arguments and return the result in Dataframe.
-
Instead of just
nrecords, you can fetch all records as well by usingdiscover.<INDEX_LEVEL_NAME>.get_all_records_as_df()
2.2.2.2.2 Querying at the Dataset Level
- Fetch dataset entries that match against the
valuefor afield.
discover.dataset_repo.get_n_datasets_for_field(field, value, regexp, n=Discover.DEFAULT_PAGE_SIZE, cursor=None, sort_by=Discover.DEFAULT_SORT_ORDER)Optionally a regular expression (regexp) can be provided instead of a value.
For example, The following query lists all datasets in the repository, where ‘publication name’ fields has pubmed id ‘28756234’
discover.dataset_repo.get_n_datasets_for_field_as_df('publication_name', '28756234', regexp=False)[0]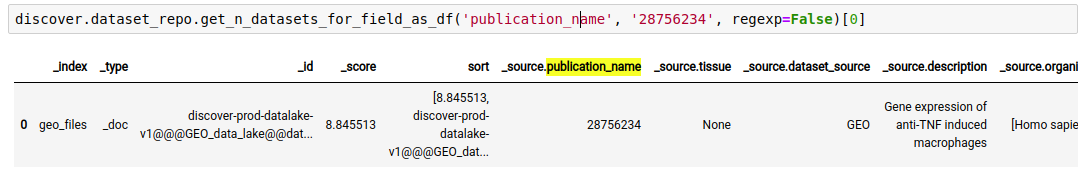
The following query lists all datasets in the repository where ‘tissue’ is ‘liver’
discover.dataset_repo.get_n_datasets_for_field_as_df('tissue', 'liver', regexp=False)[0]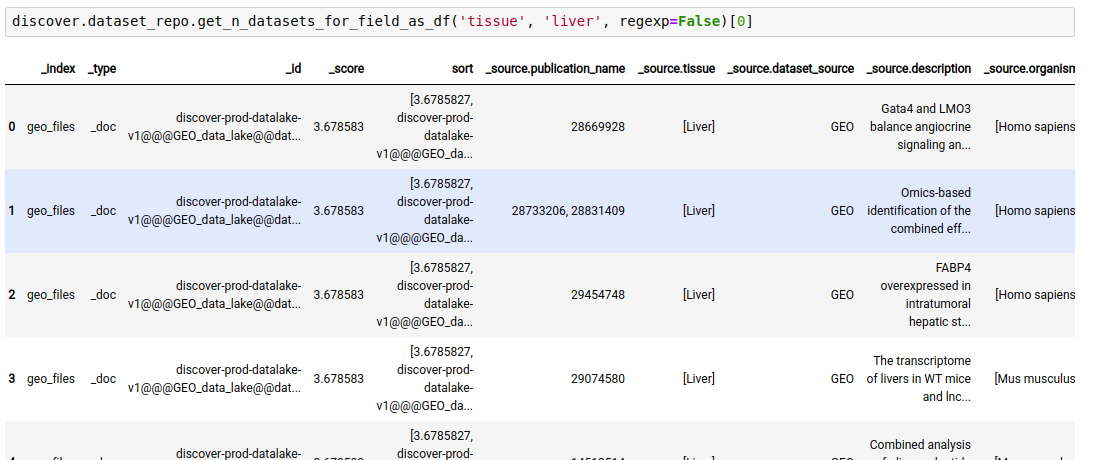
Instead of just n datasets, you can fetch all datasets as well.
discover.dataset_repo.get_all_datasets_for_field_as_df(field=None, value=None, regexp=False, sort_by=Discover.DEFAULT_SORT_ORDER)
- Fetch dataset entries that satisfy the given match combinations.
discover.dataset_repo.get_n_datasets_for_field_combination(and_fields, or_fields, not_fields, n=Discover.DEFAULT_PAGE_SIZE, cursor=None, sort_by=Discover.DEFAULT_SORT_ORDER)The following query fetches datasets where the platform is Microarray, tissue is stem cells, and the keyword ‘knockdown’ is NOT present in the description of the study.
discover.dataset_repo.get_n_datasets_for_field_combination_as_df(and_fields={"platform":"Microarray", "tissue":"stem cells"}, not_fields={"description":"knockdown"})[0]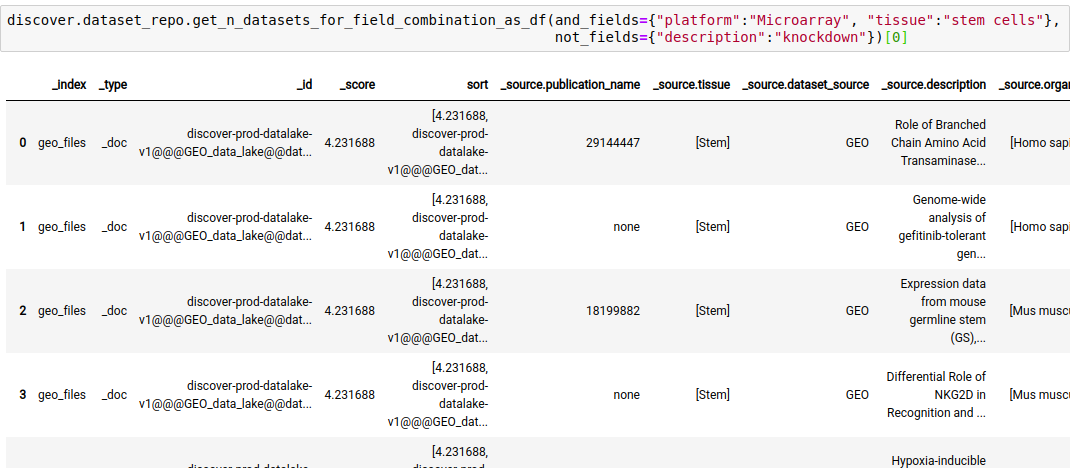
Instead of just n datasets, you can fetch all datasets as well.
discover.dataset_repo.get_all_datasets_for_field_combination_as_df(and_fields={"platform":"Microarray", "tissue":"stem cells"},not_fields={"description":"knockdown"})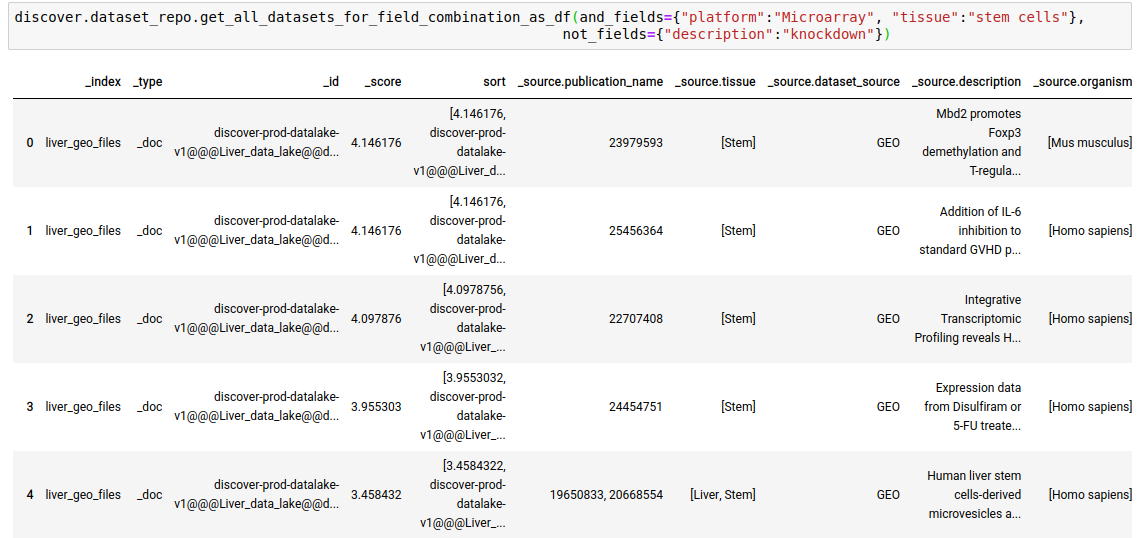
2.2.2.2.3 Querying at the feature level
- Fetch feature values matching against the
feature_nameterm. Thenhere decides the number of datasets that will be searched.
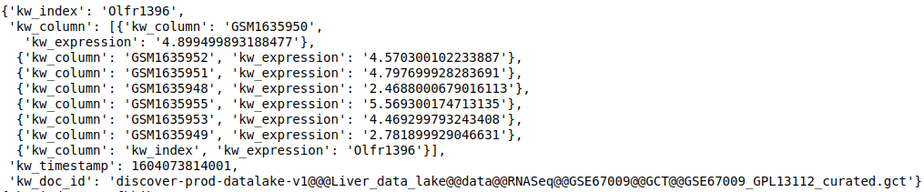
discover.feature_repo.get_n_feature_match_values(feature_name, n=Discover.DEFAULT_PAGE_SIZE, cursor=None, sort_by=Discover.DEFAULT_SORT_ORDER)The following query fetches the expression level of the gene OLfr1396 in the various samples of the repository
discover.feature_repo.get_n_feature_match_values_as_df("Olfr1396")[0]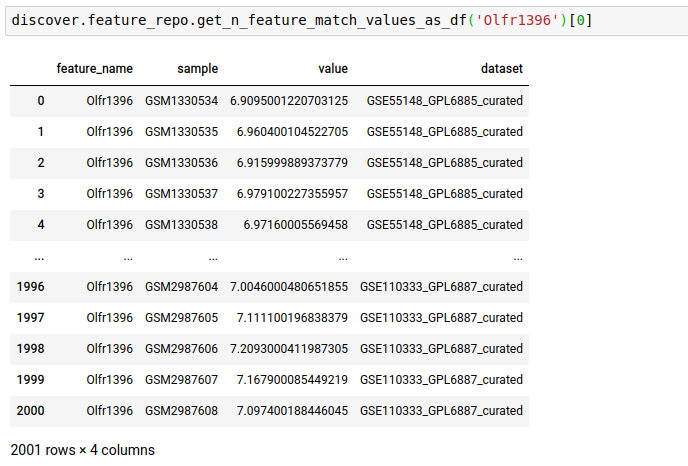
Instead of just n feature values, you can fetch all as well.
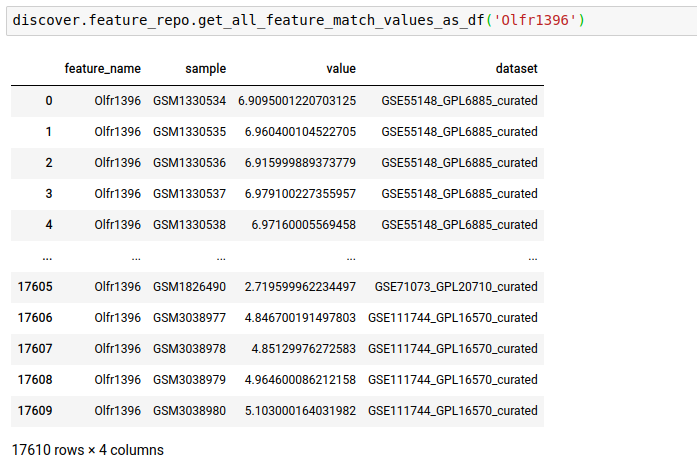
- Fetch feature values matching against the
feature_nameterm, in single-cell datasets. Thenhere decides the number of datasets that will be searched. This is for the case where the mode was set to single cell. It outputs the average expression of the gene in the pool of cells corresponding to each unique cell type in the repository, per dataset.
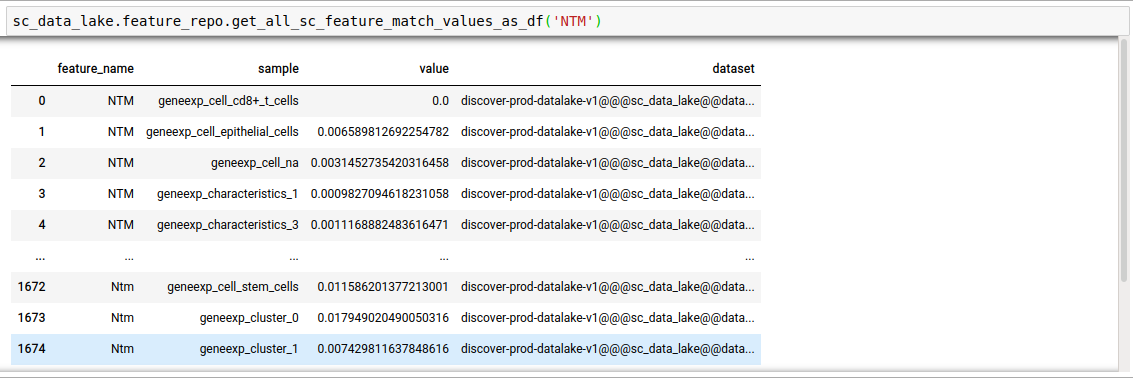
discover.feature_repo.get_n_sc_feature_match_values(feature_name, n=Discover.DEFAULT_PAGE_SIZE, cursor=None, sort_by=Discover.DEFAULT_SORT_ORDER)discover.feature_repo.get_all_sc_feature_match_values_as_df("NTM")[0]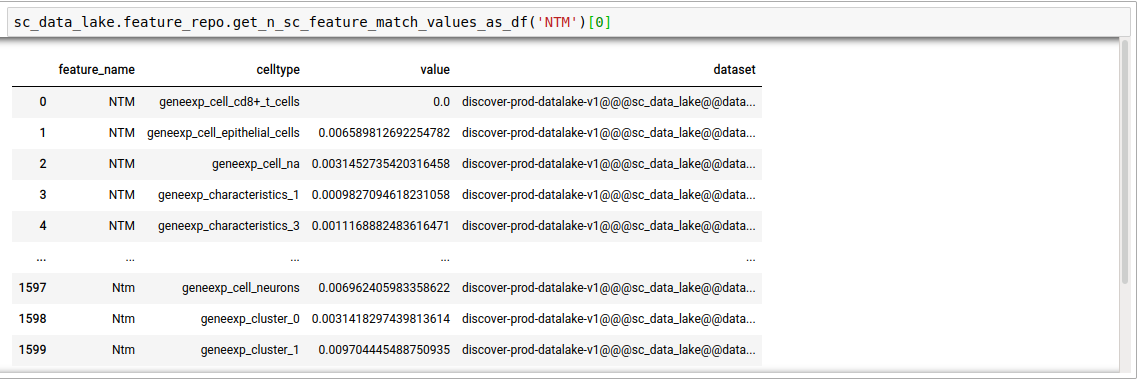
Instead of just n feature values, you can fetch all as well.
discover.feature_repo.get_all_sc_feature_match_values_as_df(feature_name, sort_by=Discover.DEFAULT_SORT_ORDER)
2.2.2.2.4 Querying at the sample level
- Fetch sample entries that match against the
valuefor afield.
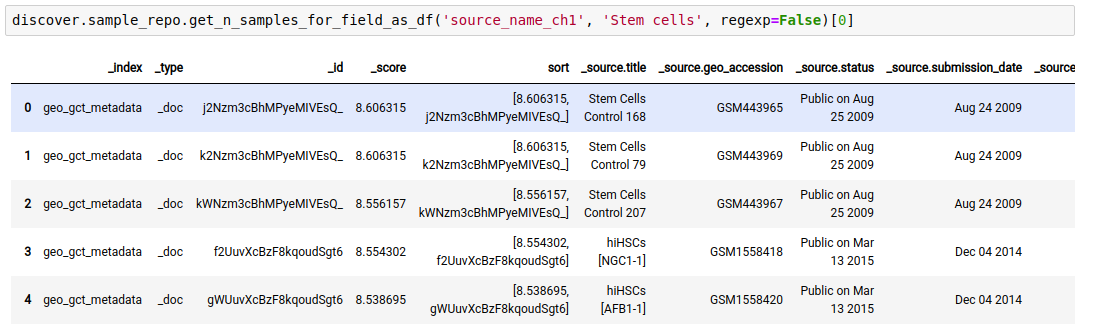
discover.sample_repo.get_n_samples_for_field(field, value, regexp, n=Discover.DEFAULT_PAGE_SIZE, cursor=None, sort_by=Discover.DEFAULT_SORT_ORDER)The following example fetches the samples whose source is “stem cells”.
- Dataframe output variant -
get_n_samples_for_field_as_df
Instead of just n feature values, you can fetch all as well.
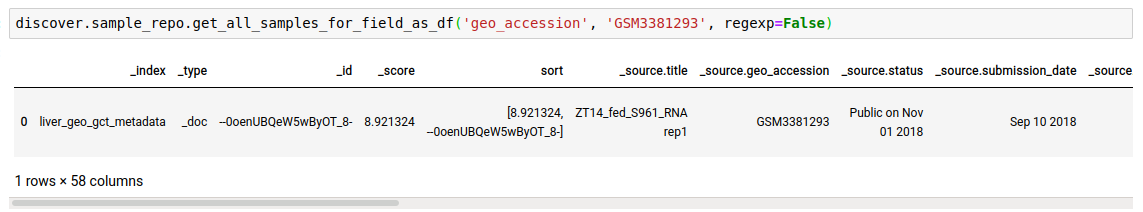
- Fetch sample entries that match against the
search_termin any of the given fields. This is useful if you do not know the field to query on beforehand or want to search across all fields, or a list of fields.
discover.sample_repo.get_n_samples_for_fields(search_term, fields=[], n=Discover.DEFAULT_PAGE_SIZE, cursor=None, sort_by=Discover.DEFAULT_SORT_ORDER)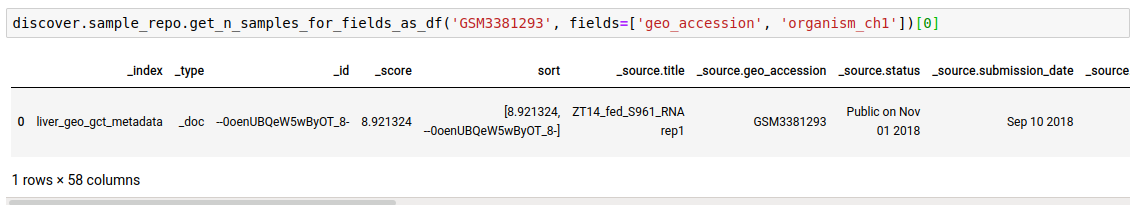
- Dataframe output variant -
get_n_samples_for_fields_as_df
Instead of just n sample matches, you can fetch all as well.
discover.sample_repo.get_all_samples_for_fields_as_df(search_term, fields=[], sort_by=Discover.DEFAULT_SORT_ORDER)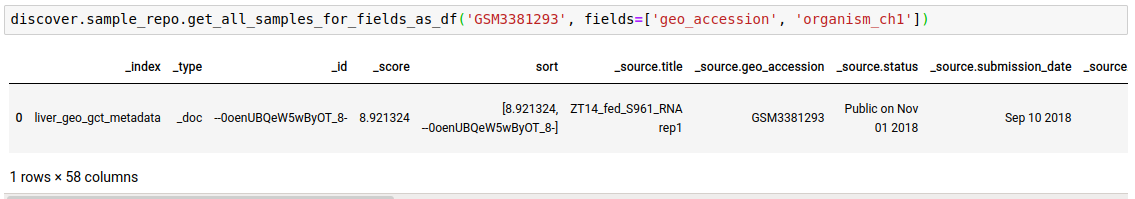
- Fetch sample entries that satisfy the given field combinations.
discover.sample_repo.get_n_samples_for_field_combinations(and_fields={}, or_fields={}, not_fields={}, n=Discover.DEFAULT_PAGE_SIZE, cursor=None, sort_by=Discover.DEFAULT_SORT_ORDER)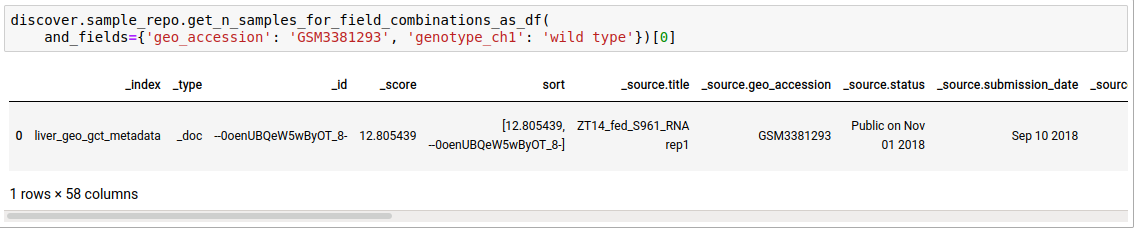
- Dataframe output variant -
get_n_samples_for_field_combinations_as_df
Instead of just n sample matches, you can fetch all as well.
discover.sample_repo.get_all_samples_for_field_combinations_as_df(and_fields={}, or_fields={}, not_fields={}, sort_by=Discover.DEFAULT_SORT_ORDER)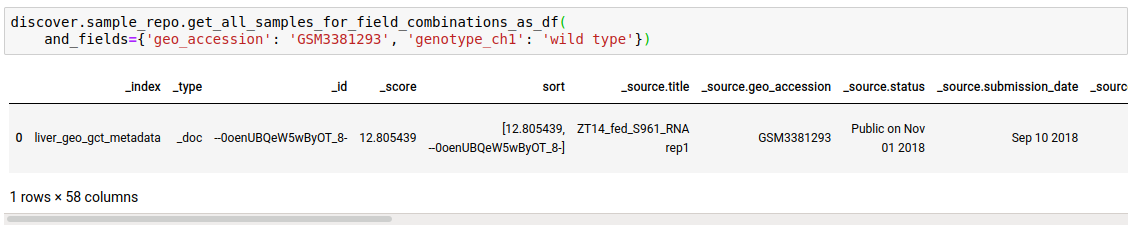
2.2.2.2.5 Access annotation repositories
The various gene annotation databases can also be accessed through discoverpy. These can be used to get information about a particular gene or a set of genes.
- Get all annotation databases
discover.annotation_repo.get_annotation_databases()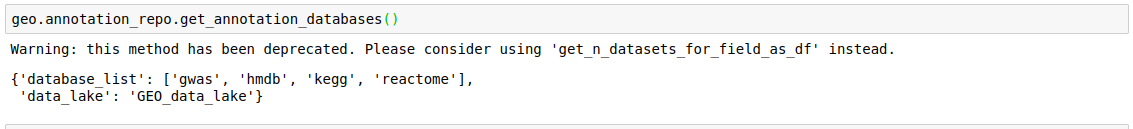
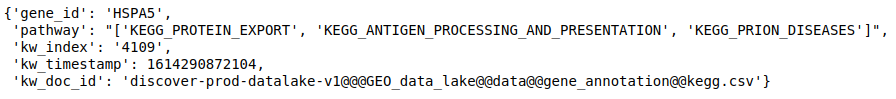
- Get annotations for a list of genes from a particular database. Getting Reactome pathways for the genes.
discover.annotation_repo.get_feature_annotation(data_lake, _docid, list_of_genes, n=10000)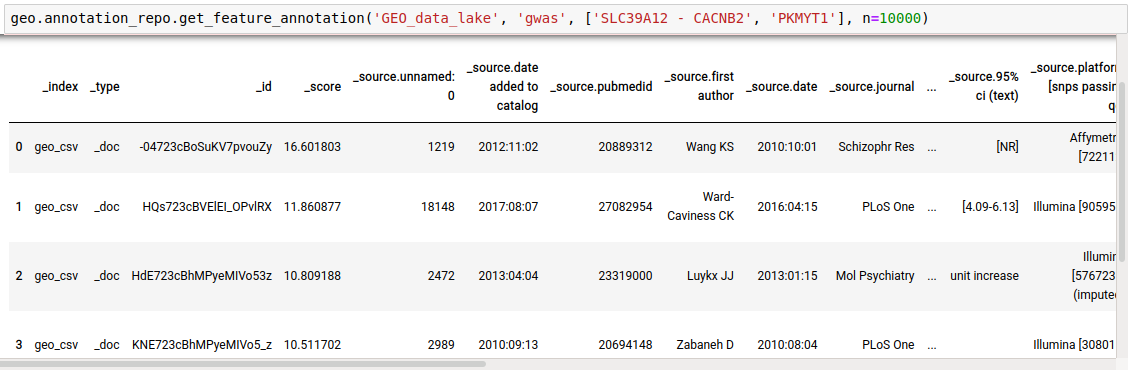
2.2.2.3 Downloading a dataset
Discoverpy can be used to download a dataset from a repository. This can be done by using the “key” of the dataset in the repository.
query_list = discover.dataset_repo.get_n_datasets_for_field('publication_name', '28756234', regexp=False)[0]
# Get key of the first file returned by the query
file_key = query_list[0]["_source"]["kw_key"]'GEO_data_lake/data/Microarray/GSE100129/GCT/GSE100129_GPL10558_curated.gct'
discover.get_file(key, repo_id="9", local_fp="downloaded_file.gct")This will download the file to the local environment.